Phonon Dispersion
Introduction
In this example, we use harmonic lattice dynamics to calculate the phonon dispersion of diamond silicon using a Tersoff potential. If you use a NEP model, we recommend you try the calorine package. It may make your life easier by significantly simplifying the process.
Importing Relevant Functions
The inputs/outputs for GPUMD are processed using the Atomic Simulation Environment (ASE) package.
[59]:
from pylab import *
from ase.build import bulk
from ase.io import write
Preparting the Inputs
The structure as specified is 64-atom diamond silicon at zero temperature and zero pressure. We use the minimal Tersoff potential [Fan 2020].
Generate the model.xyz file:
Create Si Unit Cell & Add Basis
[60]:
a=5.434
Si_UC = bulk('Si', 'diamond', a=a)
Si_UC
[60]:
Atoms(symbols='Si2', pbc=True, cell=[[0.0, 2.717, 2.717], [2.717, 0.0, 2.717], [2.717, 2.717, 0.0]])
[61]:
num_atoms = len(Si_UC)
Si_UC.unitcell = list(range(num_atoms))
Si_UC.basis = list(range(num_atoms))
Transform Si to Cubic Supercell
[62]:
# Create 8 atom diamond structure
Si = Si_UC.repeat([2,2,1])
Si.set_cell([a, a, a])
Si.wrap()
# Complete full supercell
Si = Si.repeat([2,2,2])
Si
[62]:
Atoms(symbols='Si64', pbc=True, cell=[10.868, 10.868, 10.868])
Write model.xyz File
[63]:
write("model.xyz", Si)
Write basis.in File
The basis.in file reads:
2
0 28
4 28
0
0
0
0
1
1
1
1
...
Here the primitive cell is chosen as the unit cell. There are only two basis atoms in the unit cell, as indicated by the number 2 in the first line.
The next two lines list the indices (0 and 4) and masses (both are 28 amu) for the two basis atoms.
The next lines map all the atoms (including the basis atoms) in the super cell to the basis atoms: atoms equivalent to atom 0 have a label 0, and atoms equivalent to atom 1 have a label 1.
Note: The basis.in file generated by this Jupyter notebook may look different, but the same concepts apply and the results will be the same.
[64]:
N_basis = 2
string = f"{N_basis}\n0 28\n1 28\n" + "0\n1\n" * 32
with open("basis.in", "w") as f:
f.write(string)
Write kpoints.in File
The \(k\) vectors are defined in the reciprocal space with respect to the unit cell chosen in the basis.in file.
We use the \(\Gamma-X-K-\Gamma-L\) path, with 400 \(k\) points in total.
[65]:
tol = 1e-15
npoints = 400
path = Si_UC.cell.bandpath(path='GXKGL', npoints=npoints)
b = Si_UC.cell.reciprocal() * 2 * np.pi
gpumd_kpts = np.matmul(path.kpts, b)
gpumd_kpts[np.abs(gpumd_kpts) < tol] = 0.0
np.savetxt("kpoints.in", gpumd_kpts, header=str(npoints), comments='', fmt='%g')
linear_path, sym_points, labels = path.get_linear_kpoint_axis()
The run.in file:
The run.in input file is given below:
potential ../../../potentials/tersoff/Si_Fan_2019.txt
compute_phonon 5 0.005 # in units of A
The first line with the potential keyword states that the potential to be used is specified in the file Si_Fan_2019.txt.
The second line with the compute_phonon keyword tells that the force constants will be calculated with a cutoff of 5.0 \(\mathring A\) (here the point is that first and second nearest neighbors need to be included) and a displacement of 0.005 \(\mathring A\) will be used in the finite-displacement method.
Results and Discussion
Figure Properties
[66]:
aw = 2
fs = 24
font = {'size' : fs}
matplotlib.rc('font', **font)
matplotlib.rc('axes' , linewidth=aw)
def set_fig_properties(ax_list):
tl = 8
tw = 2
tlm = 4
for ax in ax_list:
ax.tick_params(which='major', length=tl, width=tw)
ax.tick_params(which='minor', length=tlm, width=tw)
ax.tick_params(which='both', axis='both', direction='in', right=True, top=True)
Plot Phonon Dispersion
The omega2.out output file is loaded and processed to create the following figure. The previously defined kpoints are used for the \(x\)-axis.
[67]:
omega2_array = np.loadtxt("omega2.out")
nu = np.sqrt(omega2_array)/(2*np.pi)
C:\Users\hityingph\AppData\Local\Temp\ipykernel_3172\3130309619.py:2: RuntimeWarning: invalid value encountered in sqrt
nu = np.sqrt(omega2_array)/(2*np.pi)
[68]:
figure(figsize=(10,10))
set_fig_properties([gca()])
vlines(sym_points, ymin=0, ymax=17)
plot(linear_path, nu, color='C0',lw=3)
xlim([0, max(linear_path)])
gca().set_xticks(sym_points)
gca().set_xticklabels([r'$\Gamma$','X', 'K', r'$\Gamma$', 'L'])
ylim([0, 17])
ylabel(r'$\nu$ (THz)')
show()
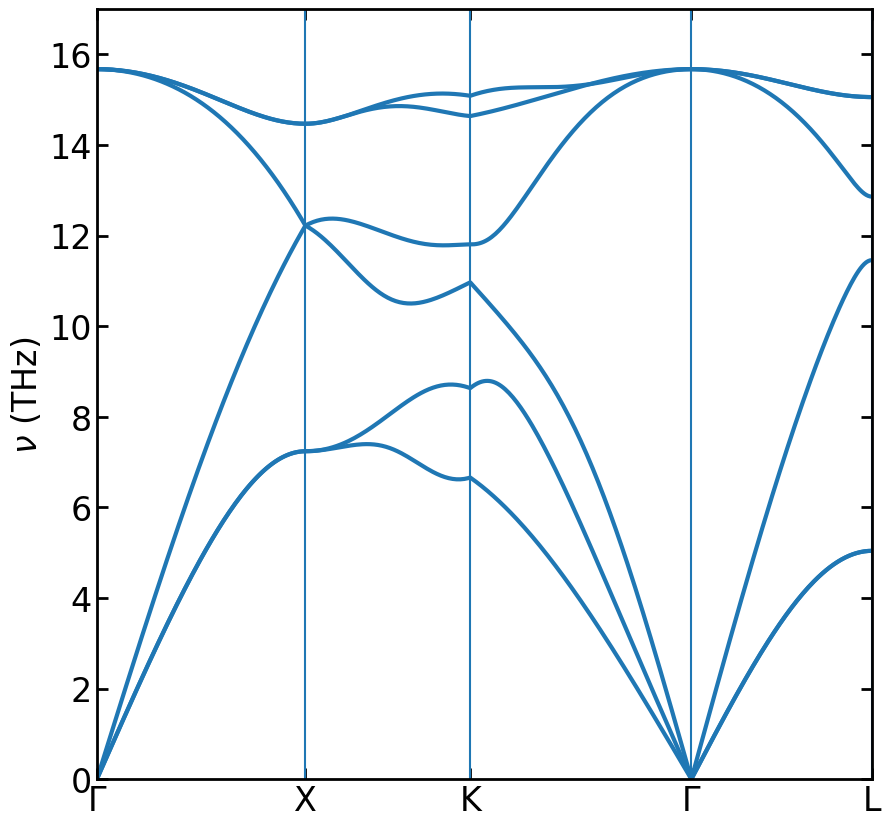
Phonon dispersion of silicon crystal described by the mini-Tersoff potential.
The above figure shows the phonon dispersion of silicon crystal described by the mini-Tersoff potential [Fan 2020]
References
[Fan 2020] Zheyong Fan, Yanzhou Wang, Xiaokun Gu, Ping Qian, Yanjing Su, and Tapio Ala-Nissila, A minimal Tersoff potential for diamond silicon with improved descriptions of elastic and phonon transport properties, J. Phys.: Condens. Matter 32 135901 (2020).